背景:
经典问题:通过质谱数据鉴别了好几个人类的、新的蛋白质序列,想探究这些所谓novel蛋白质的进化关系(也就是进行novel新蛋白质的功能注释)。
只有蛋白质序列,暂时只能通过质谱数据确认这个可能是新的蛋白质(可能性存疑),然后想探究这个novel蛋白质的进化关系,也算是验证的一个手段。
核心是:
1️⃣找到该蛋白质序列的同源序列:依据各种指标和方法
以该人类novel蛋白序列为主,通过序列相似性搜索,从最相似的物种开始,逐步扩大搜索范围,最终找到所有同源序列;
2️⃣找到了同源序列,再进行多序列比对构建进化树
第一阶段:序列确认与基本特性分析
在开始复杂的进化分析前,先彻底了解这些novel蛋白质序列的性质。
- 序列质量检查:蛋白质测序相关上游问题,质控(涉及到蛋白质组学数据分析,流程把控以及推理需要注意,因为我不是做蛋白质组学数据分析这一块的,此处默认下游衔接流程基本没问题)
- 确保蛋白质序列是从质谱数据中通过_de novo_测序和数据库搜索后唯一确定的。检查其肽段覆盖度,确保序列可信。
- 基本生物物理特性分析:
- 使用ExPASy ProtParam(https://web.expasy.org/protparam/) 工具。输入序列,可以快速获得:
- 分子量、等电点(pI)
- 氨基酸组成
- 不稳定指数(判断是否稳定)
- 脂肪指数等
- 目的:这些特性可以为后续的序列比对提供间接参考(例如,一个非常酸性的蛋白和非常碱性的蛋白可能亲缘关系较远),当然仅作为参考,因为现在在序列水平上分析一个蛋白质的理化性质的参数、工具非常多(此处只是举其中的一个工具作为例子),这里只能说对于序列的性质有一个基本的认知,分析其理化性质是一个不错的切入点。
- 使用ExPASy ProtParam(https://web.expasy.org/protparam/) 工具。输入序列,可以快速获得:
- 结构域与功能位点预测:核心
- 使用 InterProScan 或 Pfam 数据库。
- 输入序列,工具会将其与多个数据库(Pfam, SMART, PROSITE, CDD等,这里蛋白质功能结构域注释的数据库也不报菜名了,这种数据库在uniprot中关联的有很多)比对,识别其中包含的保守结构域(Domains)、motifs和功能位点。
- 目的:我个人认为这是最关键的一步! 如果蛋白含有任何已知的结构域(即使整体序列是新的),那么这个结构域就是进行后续分析的“金钥匙”。可以将分析从“整个蛋白”聚焦到“这个保守结构域”,大大增加找到远缘同源物的几率。
第二阶段:同源序列搜寻(核心步骤,多种方案)
这是回答“其他物种有没有”的问题的核心,从序列分析的角度来讲,如果起点只有序列,那么后续一切下游任务,基本上都是序列比对+参考数据库(非常传统、非常基础,但是也是非常有效的一种方法),
我个人一般是从BLAST系列(最基本的比对工具、金标准),再过渡到HMM建模的序列分析工具,只能说建议按以下顺序由敏感度低到高进行操作,仅供参考,作为个人意见。
Stage1:标准BLAST系列工具(快速、全面)
BLAST应该非常经典了,基本上所有学过生物信息学的人,在本科阶段第一次接触到序列比对算法,一定会学到BLAST(Basic Local Alignment Search Tool,基本局部比对搜索工具)。
它是一种经典且实用的序列比对工具,一般用来帮助查找某条序列(query)和已知数据库中哪些序列(reference)相似。
这种“找亲戚”的方式被广泛用于判断基因或蛋白的可能功能、物种来源或进化关系。
BLAST 不止有一种类型,它其实是一系列针对不同需求设计的工具。
核心思路是比对序列找相似的片段,但根据你输入的是 DNA 还是蛋白质序列,以及你想在哪种类型的数据库中搜索,BLAST 提供了五种常见的比对方式:

- BLASTN:用来比对核酸序列,比如 DNA 或 RNA。适用于你有一条 DNA 序列,想找出与之相似的其他 DNA。
- BLASTP:用来比对蛋白质序列。如果你有一个蛋白序列,想知道它有没有类似的已知蛋白,就用这个。
这两种是最基础、最常用的“同类型比对”工具,也就是核酸对核酸、蛋白对蛋白。适合大多数初学者日常遇到的比对需求。
而对于不同类型的比对,就需要借助翻译功能,也就是把 DNA 翻译成蛋白质来看。比如:
- BLASTX:将核苷酸序列作为查询序列,通过在六个可能的阅读框(6个ORF,正反三联体,3种可能位置x2)中对其进行翻译,生成六条假设的蛋白质序列,并将这些蛋白质序列与蛋白质数据库中的序列进行比对,用于识别可能编码的蛋白产物或功能域。
- TBLASTN:以蛋白质序列作为查询序列,将数据库中的核苷酸序列翻译为六个可能的蛋白质序列(对应不同阅读框),再将查询蛋白序列与这些翻译后的序列进行比对,用于在基因组或转录组中寻找潜在的编码区域。
- TBLASTX:将查询序列和数据库中的核苷酸序列都翻译为六个阅读框中的蛋白质序列,随后在蛋白质层面上进行比对,用于识别即使在核苷酸层面不显著但在编码蛋白层面具有同源性的序列。适用于寻找跨物种的远缘同源关系或保守功能区段。
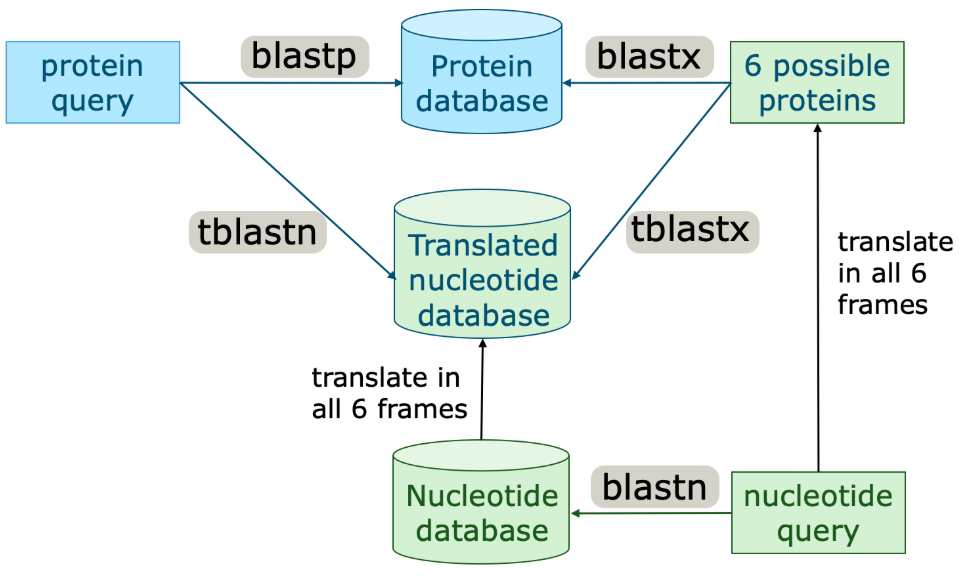
主要可以从query、reference,以及比对的level上去归类(联系与区分)这5种工具:
| BLAST 类型 | 查询序列 | 数据库序列 | 比对层级 | 常见用途 |
|---|---|---|---|---|
| blastn | 核苷酸 | 核苷酸 | 核苷酸 | 序列鉴定;适用于各类生物物种的核酸比对 |
| blastx | 核苷酸(翻译为蛋白) | 蛋白质 | 蛋白质 | 推测查询序列可能编码的蛋白质;检测潜在的新型病毒 |
| blastp | 蛋白质 | 蛋白质 | 蛋白质 | 蛋白质序列的功能注释与相似性搜索 |
| tblastx | 核苷酸(翻译为蛋白) | 核苷酸(翻译为蛋白) | 蛋白质 | 根据编码潜力在蛋白层面寻找远缘同源的核酸序列 |
| tblastn | 蛋白质 | 核苷酸(翻译为蛋白) | 蛋白质 | 查找数据库中可能编码与查询蛋白类似的核酸序列 |
- BLASTP (Protein BLAST):使用蛋白质序列查询蛋白质数据库
- 数据库:选择
Protein Data Bank proteins (pdb)或non-redundant protein sequences (nr)。nr库更全面,包含更多物种和预测蛋白,具体参考数据库看官网更新,毕竟这年头数据库三天两头就更新。 - 参数:默认即可。如果结果太多或太少,可以调整
Expect threshold (e-value),默认值0.05通常足够。可以将Max target sequences调到500或1000,以看到更多结果。——》作为参考,相关参数、意义见官网 - 目的:快速找到与蛋白序列相似度高的直系同源物(Orthologs)和旁系同源物(Paralogs)。重点关注来自非人哺乳动物(小鼠、大鼠、狗)、脊椎动物(鸡、蛙、鱼)等的匹配结果。
- 当然 tblastn我们也可以试一下,从核酸水平上找到一些有意思的序列性质,对于我们了解这条新蛋白质序列也是有帮助的
- 数据库:选择
剩下的选项,就是下拉NCBI Blast界面会在算法参数上看到的一些拓展BLAST工具
- PSI-BLAST (Position-Specific Iterative BLAST):https://www.ebi.ac.uk/jdispatcher/sss/psiblast

- 使用时机:如果标准BLASTP找到的同源物很少(例如,只找到灵长类或哺乳动物的),或者e值不够显著(如
e>0.001),说明蛋白可能比较分化。简单来说就是敏感度更高的蛋白序列与蛋白序列之间的比对,可以看作是一种更加高灵敏的Blast程序,对于发现远亲物种的相似蛋白或某个蛋白家族的新成员非常有效。 - 原理:第一轮用手头上的序列搜索,找到一些明显同源的序列;然后用这些序列构建一个“序列特征谱”(PSSM),再用这个谱(profile)进行第二轮搜索,可以找到更多更远的同源物。如此迭代3-5轮。
- 目的:发现远缘同源(Evolutionarily distant homologs)。如果迭代过程中匹配到的序列数量和多样性不断增加,说明成功。注意:要小心假阳性,特别是迭代后期匹配到一些“明星蛋白”(如泛素)时。
- DELTA-BLAST (Domain Enhanced Lookup Time Accelerated BLAST):

- 原理:DELTA-BLAST利用一个保守域数据库(CDD)来辅助搜索。它会先用序列去匹配已知结构域,然后用这个结构域信息(该结构域的PSSM profile)来增强搜索的敏感度和速度。
- 目的:强烈推荐! 特别是当蛋白含有已知结构域时,它的效果通常比PSI-BLAST更好、更快、更不易出错。
Stage2:更敏感的同源序列搜索工具
如果BLAST系列工具仍然找不到除人类以外的好结果,并不意味着没有同源物,可能只是因为序列分化太大。
因为BLAST采用的是启发式算法,而启发式算法在各种生物信息学问题中都有这么一类,所谓启发式,简单来说,就是它并不会穷尽所有可能的比对方式,只是用了一种更快的方法来找到“足够好”的比对结果,兼顾速度和准确度(在计算速度和比对准确性之间取得了很好的平衡,也就是每个生信算法经典的必品环节—trade-off环节),所以BLAST系列从算法角度讲不是最“严格”的算法。
所以,在BLAST系列“失败”之后,我们可以尝试HMM系列工具,或者其他从算法角度讲更“严格”的算法。
我们可以尝试使用HMM建模的序列工具,对于HMM的一些简单拓展了解,可以参考我之前的一篇博客,https://blog.csdn.net/weixin_62528784/article/details/150872038?spm=1001.2014.3001.5502
我在上一篇博客中提到过,说HMM是生物信息序列分析问题的万金油模型(在机器学习火起来前,也就是上个世纪~2010年左右阶段,基本上大部分序列分析相关的生物信息学问题,都可以看到HMM的身影)。
- HMMER:Biosequence analysis using profile hidden Markov Models(https://www.ebi.ac.uk/Tools/hmmer/home)
HMMER是一个软件包,提供了一类被称为隐马尔科夫模型的工具,这些工具用于制作蛋白质和DNA序列域家族概率模型(HMM profile),以及可以使用这些工具来注释新的序列。
不同于 Blast 系列工具,HMMER 使用的是集成算法(ensemble algorithms),而不是最佳对齐 (optimal alignment)。
简单来说,BLAST是通过全局或者局部的残基序列逐一比对,构建得分矩阵,再通过计算E值和P值来反应比对的显著性;
而HMMER是通过构建HMM模型,与数据库中的模型进行比较得到序列相似性结论。
- 原理:基于隐马尔可夫模型(HMM),比BLAST基于的简单序列比对要敏感得多。
- 步骤:
- a. 用序列在Pfam上搜索,如果找到了结构域,可以直接下载该结构域的HMM模型文件(
.hmm)。 - b. 或者,用序列自己构建一个HMM模型。使用工具
hmmbuild(HMMER套件的一部分)来构建。 - c. 用
hmmscan搜索Pfam数据库来验证模型,或用hmmsearch搜索整个UniProt或NCBI nr数据库。 - 相关步骤仅作为参考!毕竟工具更新换代快,只能说HMMER这个方向可以一试
- a. 用序列在Pfam上搜索,如果找到了结构域,可以直接下载该结构域的HMM模型文件(
- 目的:这是目前寻找极远缘同源物的金标准方法。能发现BLAST完全找不到的关系。
HMMER也有类似BLAST一样的系列套件,目前我所知的有支持四种工具,可使用不同的文件类型完成序列的查找
phmmer:protein sequence vs protein sequence database(与Blastp类似,使用一个蛋白质序列搜索蛋白质序列库)
hmmscan:protein sequence vs profile-HMM database(使用序列搜索HMM库)
hmmsearch:protein alignment/profile-HMM vs protein sequence database(使用HMM模型搜索序列库)
jackhmmer:iterative search vs protein sequence database(与psiBlast类似,迭代搜索蛋白质序列库,可使用上述三类文件)
- HHblits:https://toolkit.tuebingen.mpg.de/tools/hhblits
- 原理:基于HMM-HMM比对,比序列-序列(BLAST)和序列-谱(PSI-BLAST/HMMER)比对更强大。
- 使用:可以通过MPI Bioinformatics Toolkit 或 HH-suite 本地运行。
- 目的:敏感性最高,适用于“疑难”序列。
这里就不得不提一下HH-suite(https://github.com/soedinglab/hh-suite)工具包了,一般常用的核心工具是其中的HHsearch 和 HHblits。
HHblits 和HHsearch是基于隐马尔可夫模型(HMM)的强大同源搜索工具,广泛用于蛋白质结构预测、远程同源检测、注释建模等任务:
HHblits :快速构建 HMM,并进行 HMM-序列比对,适用于大规模数据集。
HHsearch:高精度的 HMM-HMM 比对,适合结构预测与注释分析。
第三阶段:多序列比对与系统发生树构建
一旦通过上述方法收集到了一批同源序列(至少20-30条,覆盖物种越广越好),就可以开始建树了。
- 序列准备:
- 从BLAST或HMMER结果中,下载匹配到的序列的FASTA文件。注意筛选:
- 选择代表性物种(如人、黑猩猩、小鼠、鸡、青蛙、斑马鱼、果蝇、线虫、酵母、植物等),尽量覆盖进化树的主要分支。
- 避免选择过多非常相似的序列(例如,不要选100种不同的细菌,选几个代表即可),以免树失衡。
- 如果找到了旁系同源物(同一物种内有多个拷贝),需要全部保留,以分析基因复制事件。
- 从BLAST或HMMER结果中,下载匹配到的序列的FASTA文件。注意筛选:
下面就是正常的多序列比对构建系统发生树的过程了,使用工具也只是仅供参考! 还是那句话,只是调包调工具的话,世界上有很多轮子,不分高下,要按照自己的目的去筛选想要的工具。
- 多序列比对:
- 工具:MAFFT 或 Clustal Omega等(仅参考)。MAFFT通常对全局比对更准确,推荐使用。
- 在线使用:EBI网站提供了非常方便的在线MAFFT工具。
- 目的:将所有的序列按照氨基酸的相似性进行排列,这是建树的基础。比对质量直接影响树的质量。
- 比对结果修剪:
- 工具:TrimAl 或 Gblocks等(仅参考)。
- 目的:自动修剪掉比对结果中排列质量很差、充满空位(gap)的区域,只保留保守性好、可信度高的区块用于建树。
- 构建系统发生树:
- 方法选择:
- 最大似然法:最常用、最可靠的方法。推荐工具 IQ-TREE 或 PhyML等(仅参考)。
- IQ-TREE 非常强大且易用,它会自动选择最合适的氨基酸替代模型。
- 贝叶斯推断法:结果也很可靠,但计算更耗时。工具如 MrBayes。
- 最大似然法:最常用、最可靠的方法。推荐工具 IQ-TREE 或 PhyML等(仅参考)。
- 目的:得到一棵树,其中分支的长度代表了遗传距离,节点的支持率(bootstrap值或后验概率)代表了节点的可靠性(通常>70%或>0.95认为可靠)。
- 方法选择:
- 树的可视化与解读:
- 工具:FigTree, iTOL等(仅参考)。
- 解读:
- 观察该蛋白在进化树中的分布。它只存在于脊椎动物?后生动物?还是真核生物普遍存在?甚至原核生物也有?
- 树的根部在哪里?这可以揭示起源时间。如果包含了远缘的 outgroup(如,分析真核蛋白时加入细菌或古菌序列),树根通常会落在真核和原核分支之间。
- 寻找共有的基因复制事件。例如,如果所有脊椎动物的序列都聚成两个明确的簇,说明在脊椎动物祖先中发生了一次全基因组复制事件。
第四阶段:高级分析与验证
- 共线性分析:
- 如果蛋白只在某些特定物种中存在/缺失,可以检查其基因组位置上下游的基因。如果 across 多个物种,该区域都存在一组保守的基因,那么这个区域就是一个共线性区块。这可以强力佐证系统发生分析结果,并帮助判断是同源物还是通过水平基因转移(HGT)得来的。
- 合成性分析:
- 检查与蛋白相互作用的伴侣蛋白(如果已知)在其他物种中的存在情况。如果蛋白和其伴侣蛋白在所有物种中都“同在”或“同不在”,这为蛋白的功能和进化提供了极好的旁证。
- 这一部分主要是依靠我们的生物学domain知识了,进化学得深不深、遗传深不深,当然牵涉到具体生物学背景解读和理解就有很多东西可以发散了,这里就只是简单提两嘴。